NTU_Taiwan
Experiment

Materials and Methods
DNA Origami Assembly
The triangular DNA origami was designed with the software-tool caDNAno.
The structure was assembled from 200 staple strands, using the 7249-nt long M13mp18 genome as a
scaffold. The original staple strands were first dissolved in deionized water at 100 μM concentrations,
and then mixed at equal concentrations to yield the staple mixture. With 200 staple strand solution
mixed, the final concentration of the staple mixture was 500 nM. This stock solution was stored at
-20 ℃. In addition, the other 7 staple strands were modified with functional DNA fragments. Three
of them were modified with Aptamer CD63, targeting NK92-derived exosome, with sequence of 5'
CACCCCACCTCGCTCCCGTGACACTAATGCTA 3' (Tang et al., 2023) added to the end of the staple sequence.
Another three of them were modified with MUC1 DNA Aptamer S2.2 with sequence of
5' GCAGTTGATCCTTTGGATACCCTGG 3' (Shahrad et al., 2022) synthesized to the end of the staple strand.
The last one was a DNA oligonucleotide sequence designed for sequential-pull down purification by
magnetic beads, with a capture region 5' ACTTTAATCTCACAACGGAA 3' and a 18 nt spacer between the
capture region and the origami staple (Ye et al., 2021). All of the seven staple strands with
functional DNA sequences listed in Table 1.
Assembly was performed at a 10-fold excess of staples to scaffolds in folding buffer containing 1×
TAE (Tris Acetate-EDTA) and 10 mM MgCl2 (Table 2). The annealing was carried out in a T100TM Thermal
Cycler (BIO-RAD) by heating the sample to 80 ℃ and gradually cooling it to room temperature over a
time course of 90 minutes by lower 0.6℃ per cycle (Kielar et al., 2019). Subsequently, the
functional DNA fragments were added at the same molar concentration and volume as the staple mixture,
and then incubated with the pre-folded structure at 37°C for 2 hours.
| Name | Sequences | |||
| Purification strand (P) |
GAACGGTAATCGTAAAACTAGCATGTCAATCA TACACTTACACTTACACT ACTTTAATCTCACAACGGAA |
|||
| NK92 exosome aptamer 1 (Eapta1) |
GAGAGACTATAAAGCCAACGCTCAAACAACGC TTTTT CACCCCACCTCGCTCCCGTGACACTAATGCTA |
|||
| NK92 exosome aptamer 2 (Eapta2) |
GCAGCAAGGGGGGTAATAGTAAAAAAGAAGTT TTTTT CACCCCACCTCGCTCCCGTGACACTAATGCTA |
|||
| NK92 exosome aptamer 3 (Eapta3) |
CCCAATAAATCTTACCGAAGCCCTGGAATA TTTTT CACCCCACCTCGCTCCCGTGACACTAATGCTA |
|||
| A549 cell aptamer 1 (Capta1) |
GCTGAGAGAGGAATTGAGGAAGGTTTGTTTGGATTATACT TTTTT GCAGTTGATCCTTTGGATACCCTGG |
|||
| A549 cell aptamer 2 (Capta2) |
TGATGGTGAACAGCTGATTGCCCTGCCTAATG TTTTT GCAGTTGATCCTTTGGATACCCTGG |
|||
| A549 cell aptamer 3 (Capta3) |
AGAATAGAAGACAGCATCGGAACGTAAAGA TTTTT GCAGTTGATCCTTTGGATACCCTGG |
|||
| Total volume: 100 μL | ||||
| Staple | ||||
| 500 nM staple mixture (to 30 nM) | 6 | |||
| Scaffold | ||||
| 100 nM M13mp18 scaffold (to 3 nM) | 3 | |||
| Buffer | ||||
| 10×TAE (to 1×) | 10 | |||
| 50 mM MgCl2 (to 10 mM) | 20 | |||
| ddH2O | 61 | |||
| Annealing condition: 80 ℃ 5 min → -0.6 ℃/min → 25 ℃ | ||||
DNA Origami Purification
The folded samples were purified of excess staples with 1×TAE and 10 mM MgCl2 washing buffer (the same as the DNA origami folding buffer) by spinning filtering using Amicon Ultra-0.5 mL Centrifugal Filters with 100 kDa molecular weight cut-off (Merck Millipore) (Kielar et al., 2019). Firstly, to rinse an Amicon Ultra-0.5 mL Centrifugal Filter device, apply 500 μL washing buffer into the Amicon tube, close the lid, and spin at 4,500 g at 4 ℃ for 7 min. Next, for washing the samples, load 100 μL samples and 400 μL washing buffer to an Amicon Ultra-0.5 mL Centrifugal Filter device and then spin at 4,500 g at 4 ℃ for 8 min. Wash the left three more times with 450 μL washing buffer, spinning at 4,500 g at 4 ℃ for 8 min. Finally, to recover the concentrated solute, place the Amicon Ultra-0.5 mL Centrifugal Filter device upside down in a clean microcentrifuge tube, spinning for 5 min at 4,500 g at 4 ℃ to transfer the concentrated sample from the device to the tube.
Agarose Gel Electrophoresis
Electrophoresis of the folded DNA origami sample was carried out in 1% agarose gels (22 mL 1× TAE and 0.2 g agarose powder) with electrophoresis buffer (1× TAE). The gels contained 2 μL ethidium bromide (EtBr). Load 10 μL of a sample, previously dyed by 1 μL loading dye, into a well and the samples were electrophoresed for about one hour at a half voltage.
TEM Imaging
Prepare the TEM sample a day before TEM imaging. To prepare a TEM sample, the carbon-coated TEM grids were firstly undergone glow-discharge in the Hydrophilic Treatment Apparatus. Then, drop 10 μL 20-40 nM DNA origami samples on the light side of a grid for 2 min. Liquids were removed by using filter papers and the grid was inverted on a drop of 70 μL ddH2O for 30 sec, and then moved to the other drop of 70 μL ddH2O again for 30 sec. After that, the grid was inverted on a 70 μL 2% uranyl acetate (UA) droplet for 1 min. Liquids were removed by filter papers and the grid was airdried. The grids were restored to the box for further observation. The TEM samples were examined using a transmission electron microscope (FEI Tecnai G2 F20 S-TWIN).
Sequential Pull-Down Purification
The following steps were based on Ye et al. (2021). A set of DNA oligonucleotides were used,
including capture strands, competitor strands, and release strands (Table 3).
Washing beads: 25 μL streptavidin-coated magnetic beads (BioSmart) were transferred to a
tube and washed with 250 μL washing buffer (WB, 10 mM Tris–HCl, 1 mM EDTA, 12 mM MgCl2,
5 mM NaCl, and 0.05% Tween 20) by vortexing for 30 sec. Leave the tube at 25℃ for 1-3 mins and place it on the magnetic
rack for 1-3 mins and remove the supernatant. Repeat the washing step two more times.
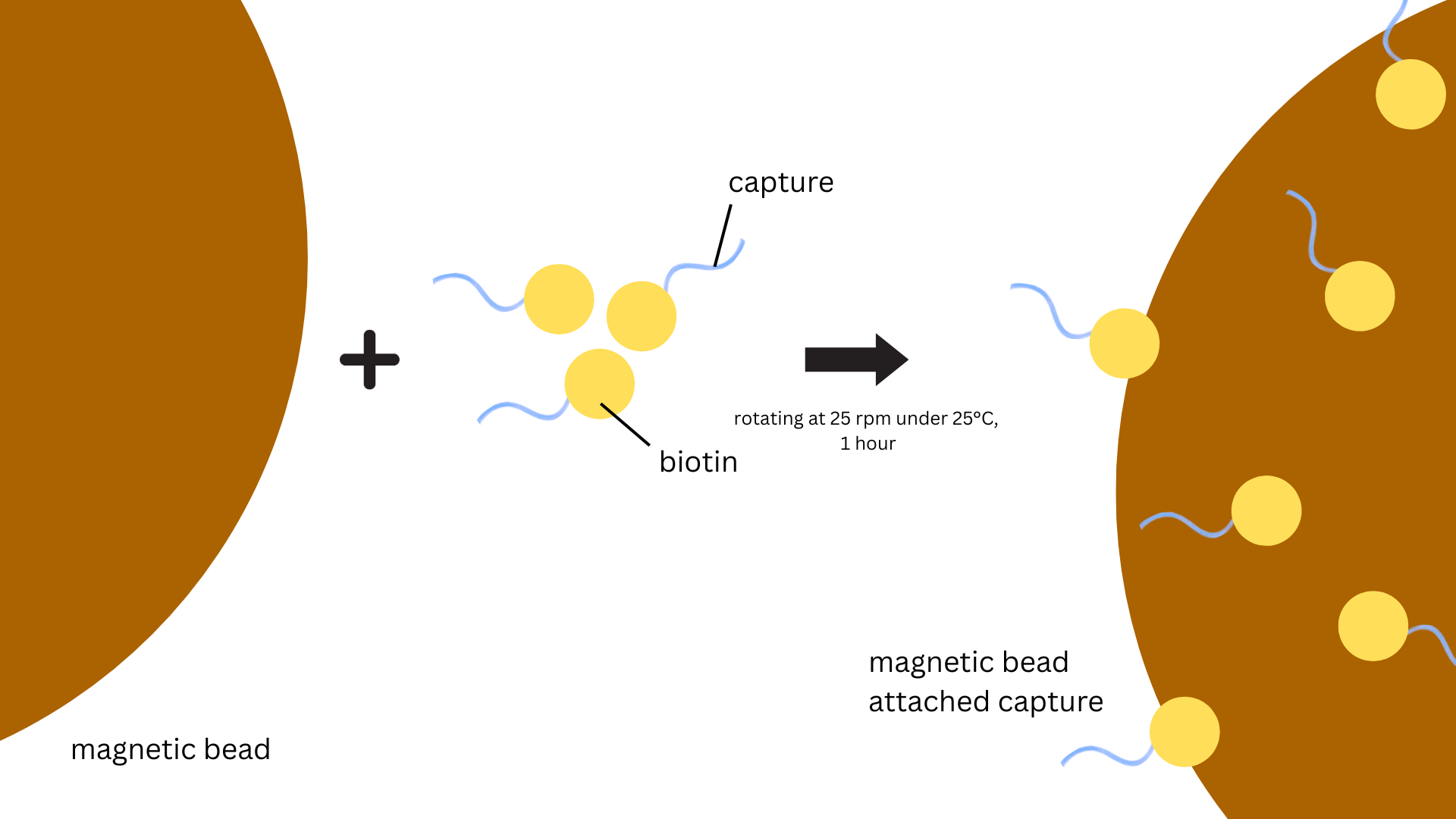
Capture procedure:
Capture strands were dissolved in deionized water and adjusted the concentration to 1 μM.
100 μL 1μM capture strand solution was added to the washed magnetic beads. The mixture was
incubated for 1 h at room temperature while kept under constant rotation at 25 rpm
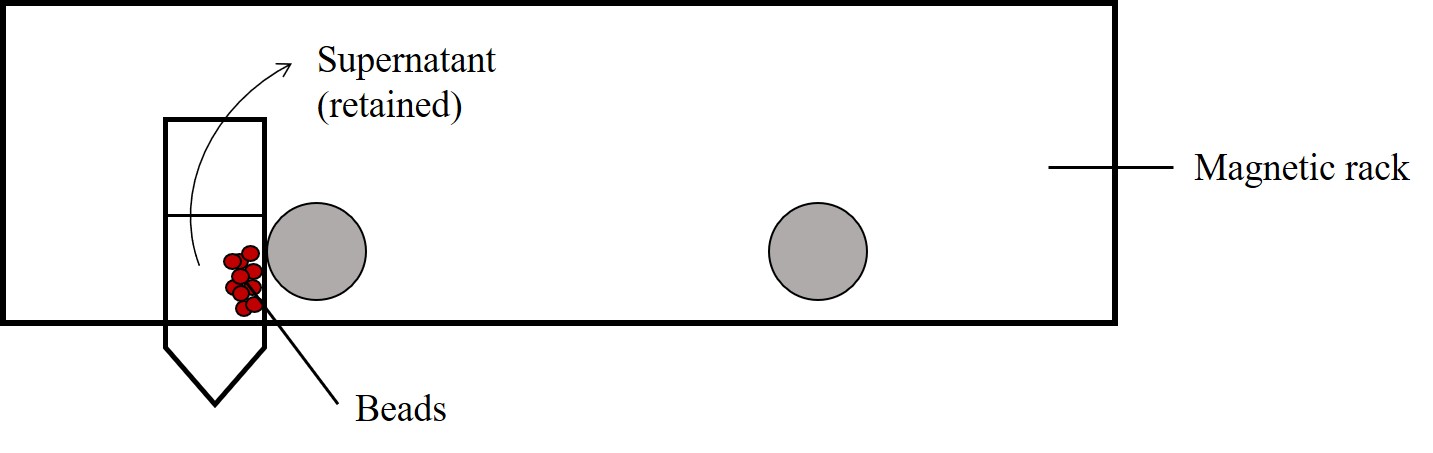
(OH-800D, Yihder) (Figure 1).
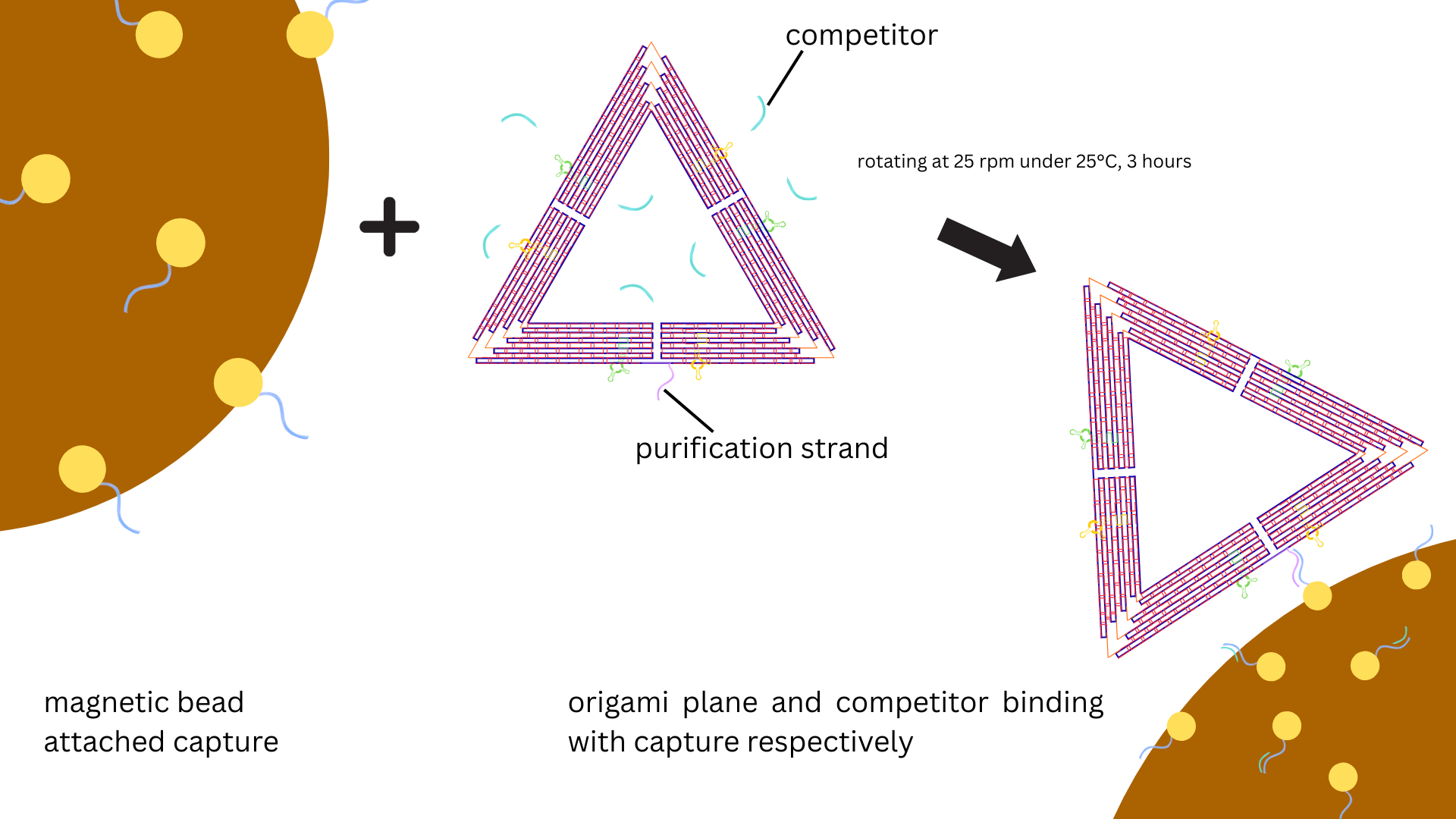
The coated beads were washed again three times with 250 µL WB to remove the unbound capture
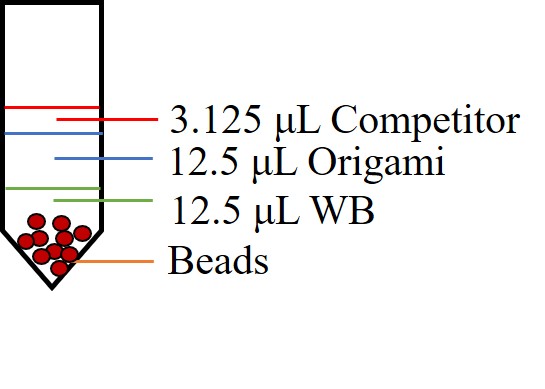
oligonucleotides. The washed beads were resuspended in 12.5 μL WB with 12.5 μL 20 nM DNA origami
samples. 3.125 μL 2μM competitor strand solution was also added, a ratio of 1:8 between competitor
and capture strands (Figure 4). The mixture of capture strand-coated beads and DNA origami and
competitor strands was incubated for 3 h at room temperature at constant rotation at 25 rpm
(OH-800D, Yihder) (Figure 2). The beads were then pelleted by using the magnetic rack and the
supernatant was sucked out for agarose gel electrophoresis analysis (Figure 5). The beads were
then washed three times with 250 μL WB.
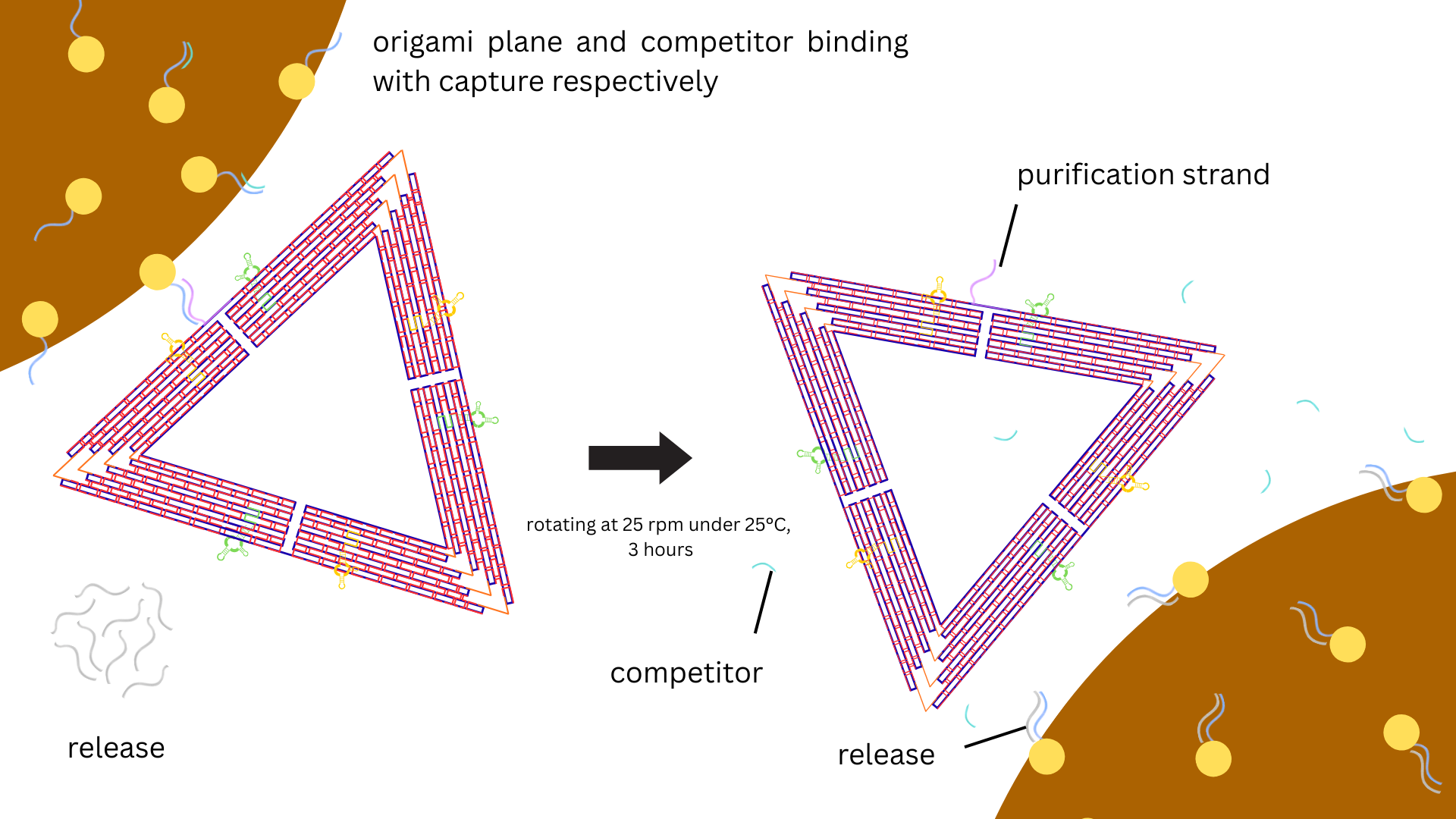
Release procedure:
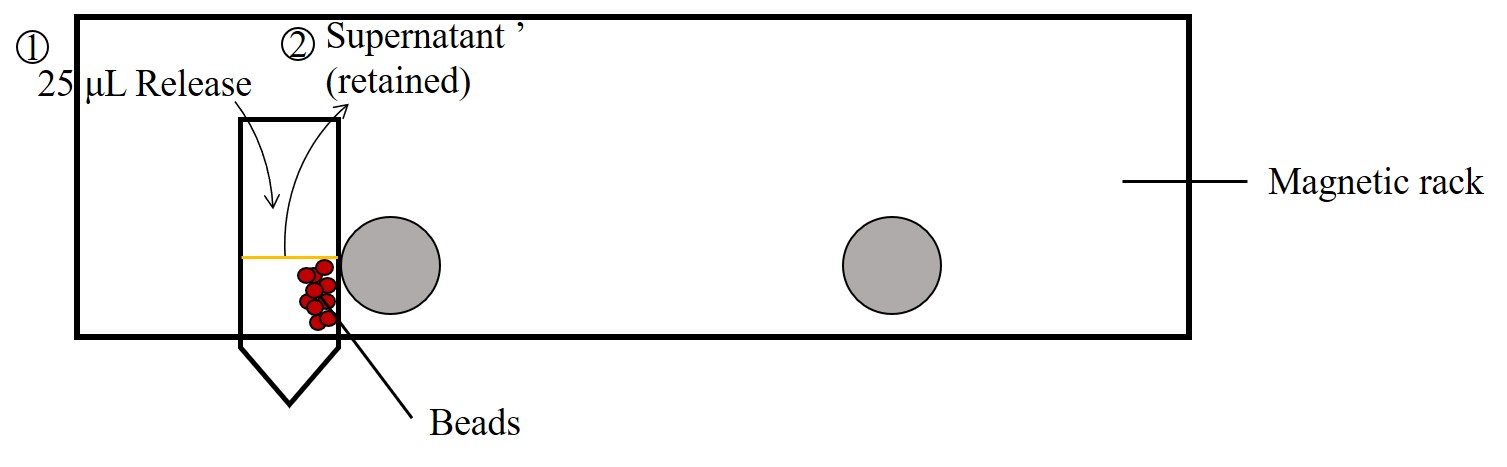
To release the DNA origami from the capture beads, 4 μM release strand solution (in 10 mM Tris–HCl,
1 mM EDTA, 6 mM MgCl2, and 300 mM NaCl) was added to the washed and
pelleted beads at the same volume as the applied bead stock solution. The mixture was incubated for
3 h at room temperature at constant rotation at 25 rpm (OH-800D, Yihder) (Figure 3).
Beads were then pelleted by the magnetic rack and the supernatant with release strands was analyzed
by agarose gel electrophoresis (Figure 6).
| Capture | Biotin-CGATGCATGATCTAGTTCCGTTGTGAGATTAAAGT | |||
| Competitor | ACTTTAATCTCACAACGGAA | |||
| Release | ACTTTAATCTCACAACGGAA CTAGATCATGCATCG | |||
Materials and Methods
DNA Origami Assembly and Purification Checked by Gel Electrophoresis
From lane 2 to lane 7, scaffolds with different preservation time were tested. The gel electrophoresis result has shown that all of the folded scaffolds were still in a good situation for further DNA origami assembly because each of them had a clear band that lined in a line (Figure 7). From lane 8 to 10, the same staple samples with different folded or purified condition were tested. For the unpurified staple samples, bands could clearly be seen at the bottom, while the band was nearly disappeared after the staple sample was purified (Figure 7). This suggested that Amicon Ultra-0.5 mL Centrifugal Filters with 100 kDa MWCO could successfully purify excess staple strands. From lane 11 to 13, DNA origami assembly was tested. Compare the unfolded DNA sample with the folded DNA origami samples, the upper bands of the folded ones were seen to have a downward shift (Figure 7). This suggested that the change of DNA structure might occur, implying a possible DNA origami structure assembly. In this group, we could also see bands at the bottom of the unpurified samples while the band at the bottom of the purified DNA origami samples disappeared. The result of gel electrophoresis showed that we possibly completed DNA origami folding and our purification methods could successfully remove excess staple strands, hence gaining the higher purity of DNA origami sample. To confirm the DNA origami structure, we further took TEM imaging.
| Lane | Sample name | Folded | Purified | |
| 1 | 1kb ladder | - | - | |
| 2 | (5/20) Sc | X | X | |
| 3 | (5/20) Sc | O | X | |
| 4 | (1/20) Sc | O | O | |
| 5 | (3/20) Sc | O | O | |
| 6 | (4/20) Sc | O | O | |
| 7 | (5/20) Sc | O | O | |
| 8 | Staples (St) | X | X | |
| 9 | St | O | X | |
| 10 | St | O | O | |
| 11 | Sc+St | X | X | |
| 12 | Sc+St | O | X | |
| 13 | Sc+St | O | O | |
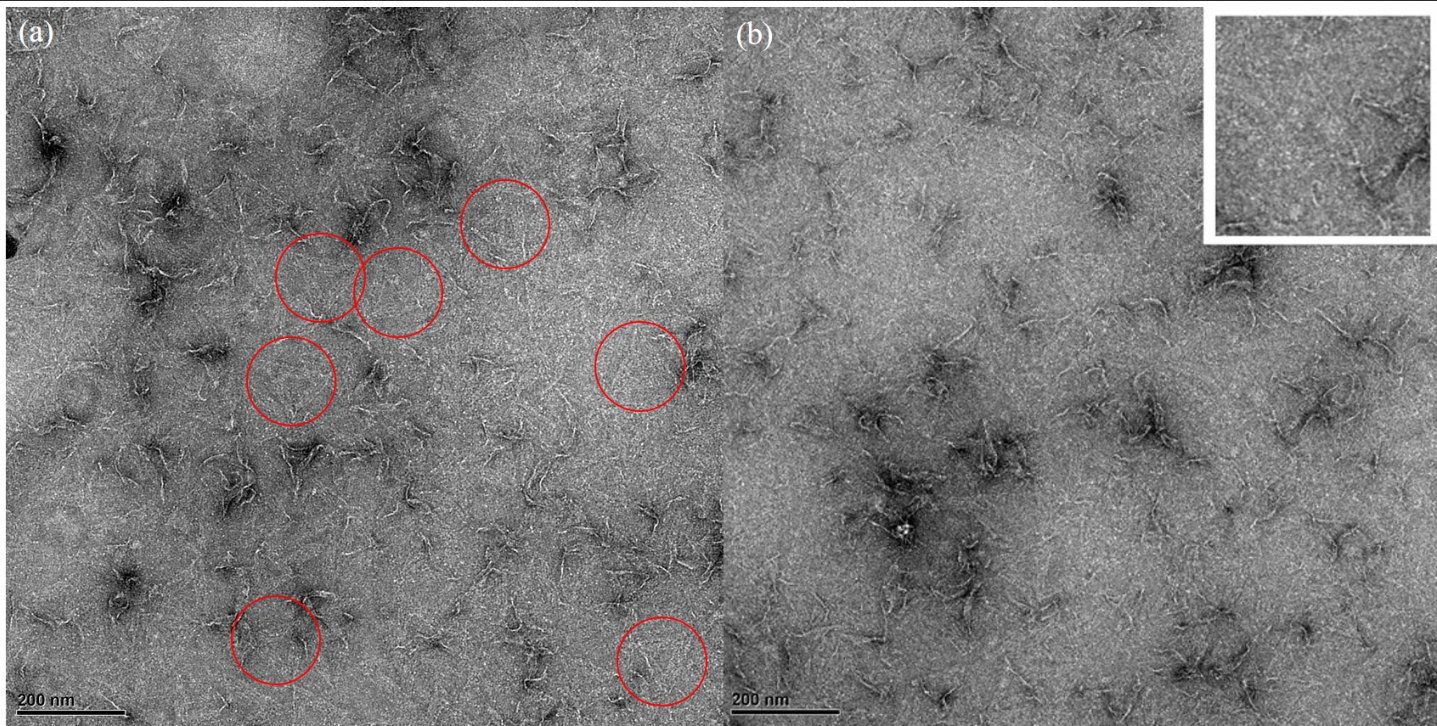
TEM imaging
30 nM DNA origami samples were fixed on the TEM grids with 2 % uranyl acetate (UA) negatively-stained. The grids were later examined by TEM under 15000× (Figure 8). Several triangular DNA origami nanostructures were seen. The DNA origami structure was shown in an intact planer equilateral triangle (Figure 5-red circle). The length of each side of the triangle was approximately 120 nm.
Sequential pull-down purification by magnetic beads
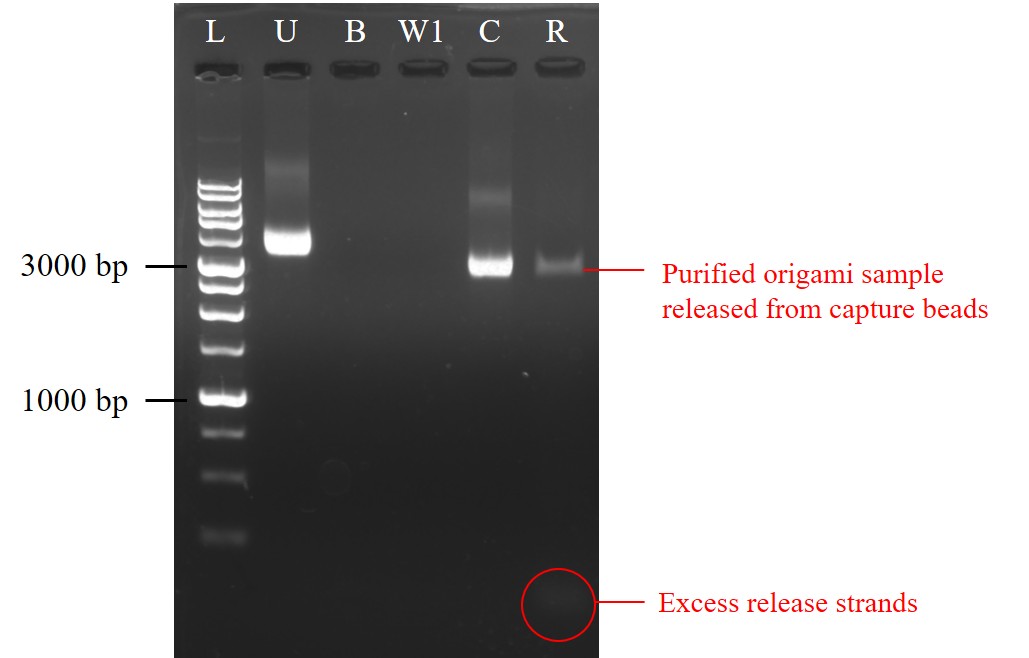
Biotin-modified capture strands were first bound to streptavidin-coated magnetic beads. After capture strands binding to the beads, the supernatant was collected (B) and analyzed by agarose gel electrophoresis (AGE). The beads were then washed and the supernatant the first-time wash was also collected (W1). The AGE result showed that nearly no bands could be seen, indicating capture strands might almost bound on the beads (Figure 9). Later, DNA origami samples were added to the capture-coated beads. After incubation, the supernatant was collected (C) for AGE analysis in order to check whether origami was captured by beads. However, a bright band was shown in the sample C, which meant some DNA origami had not been captured through the capture step (Figure 9). While compare with 10 nM DNA origami sample without carrying out sequential pull-down purification (U), which was the same molar concentration as the origami sample added in the capture step, the sample C had a flatter and a little bit darker band than the sample U, showing decrease of DNA origami; in other words, some DNA origami had been captured by beads. Finally, release strand solution was added and the supernatant was collected after incubation (R) to see whether DNA origami was released from the capture beads. From the result of AGE, a dark but still clear band could be seen in the sample R (Figure 9). This suggested there were DNA origami samples released from capture beads, ensuring that some DNA origami was truly bound on the beads in the capture step and the purified DNA origami sample was gained after the release step. Though the recovery yield of DNA origami could still be improved, we got a preliminary success in sequential pull-down purification. This purification method could further be used to purify NK-92 cell exosome by our DNA origami with functional aptamers specifically binding to exosome and then purifying through sequential pull-down reactions.
| Name | Sample content | |||
| L | 1kb DNA ladder | |||
| U | 10nM DNA origami sample | |||
| B | Supernatant after the step of capture strands binding to beads | |||
| W1 | Supernatant of the first time washing the capture-coated beads | |||
| C | Supernatant after the step of origami being captured | |||
| R | Supernatant after the release step | |||
Reference
-
Kielar, C., Xin, Y., Xu, X., Zhu, S., Gorin, N., Grundmeier, G., Möser, C., Smith, D. M., & Keller, A. (2019).
Effect of Staple Age on DNA Origami Nanostructure Assembly and Stability.
Molecules, 24(14), 2577.
https://www.mdpi.com/1420-3049/24/14/2577 -
Shahrad, S., Rajabi, M., Javadi, H., Karimi Zarchi, A. A., & Darvishi, M. H. (2022).
Targeting lung cancer cells with MUC1 aptamer-functionalized PLA-PEG nanocarriers.
Sci Rep, 12(1), 4718.
https://doi.org/10.1038/s41598-022-08759-z -
Tang, J., Jia, X., Li, Q., Cui, Z., Liang, A., Ke, B., Yang, D., & Yao, C. (2023).
A DNA-based hydrogel for exosome separation and biomedical applications.
Proceedings of the National Academy of Sciences, 120(28), e2303822120.
https://doi.org/doi:10.1073/pnas.2303822120 -
Ye, J., Teske, J., Kemper, U., & Seidel, R. (2021). Sequential Pull‐Down Purification of DNA Origami Superstructures.
Small, 17(17), 2007218.
https://doi.org/10.1002/smll.202007218